Nutrizionista Cosenza
Dott.ssa Chiara Palermo
Consigli per una sana e corretta alimentazione, lezioni di educazione alimentare
Nutrizione clinica, nutrizione sportiva, dieta chetogenetica
Benvenuti
Cerchi un nutrizionista a Cosenza e provincia?
La Dott.ssa Chiara Palermo, specializzata in nutrizione clinica e nutrizione sportiva, riceve a Cosenza presso Studi Medici CS1 e a Mirto-Crosia presso Studio Nutrizionale PalMed.

Risultati duraturi nel tempo
Dieta personalizzata
Risultati veloci

Nutrizionista Cosenza - Dott.ssa Chiara Palermo

Sarò lieta di accoglierti nel mio studio per iniziare insieme un percorso di educazione alimentare che ti permetta, in maniera personalizzata, di raggiungere gli obiettivi desiderati, senza troppe rinunce a tavola, e che questi siano duraturi nel tempo.

Nozioni di educazione alimentare
Andiamo a chiarire alcuni dubbi o… vediamo qual è la risposta giusta.

L’alimentazione rappresenta un ottimo mezzo per la prevenzione e il controllo di numerose patologie. Un’alimentazione corretta e personalizzata ti servirà per dare il meglio di te in tutte le attività giornaliere sia fisiche che mentali.
Endocrinologia
Gastroenterologia
Dislipidemie
Otorinolaringoiatria
Sistema immunitario

L’alimentazione cambia in base al genere, all’età e all’attività dei nostri ormoni. Cambiano le richieste dei diversi nutrienti….
Gravidanza e allattamento
Crescita bambini / adolescenti
Terza età

L’alimentazione preallenamento, durante l’allenamento e post allenamento è fondamentale per sfruttare al meglio l’attività fisica e raggiungere i risultati prefissati.

La chetosi è un meccanismo fisiologico alternativo dell’organismo che, se adottato in maniera controllata e in alcune condizioni, rappresenta un valido strumento per perdere peso in poco tempo e in salute. In una prima fase potrai perdere il 10% del tuo peso corporeo.
Anche i piccoli progressi sono un progresso
Non importa quanto piccoli siano i tuoi progressi, è sempre un progresso. Inizia la tua vita sana da oggi.
Dott.ssa Chiara Palermo
La tua nutrizionista a Cosenza e provincia
Il cibo è un piacere e uno dei nostri migliori alleati per stare bene, basta sapere come usarlo a tuo vantaggio.
Mangiare bene, anzi benissimo, in un modo adatto a te, alle tue esigenze ed ai tuoi gusti, consente di migliorare la tua qualità di vita.
L’alimentazione studiata per te dal Nutrizionista è uno strumento fondamentale per favorire la tua salute, il tuo benessere, il tuo buon umore
Alcuni cibi e combinazioni di alimenti ti fanno sentire più attivo, più motivato, più allegro. Favoriscono l’attenzione e la concentrazione, migliorano il rendimento nello studio, nello sport e sul lavoro. Inoltre sentirsi in forma, stare bene nel proprio corpo, piacersi, favorisce il buon umore.
Al contrario diete, combinazioni e schemi alimentari inadatti possono influenzare negativamente l’umore rendendo tristi oppure irritabili o entrambi. Una alimentazione inadatta ti può ridurre la concentrazione e l’attenzione.
Con il cibo si possono prevenire e curare alcune malattie e tanti disturbi, dal colesterolo alto alla colite, e migliorarne molte altre.
La tua salute comprende anche un peso adeguato a te.
Il controllo del peso e la linea fanno parte del benessere.
Nutrizionista Cosenza Dott.ssa Chiara Palermo ti fa scoprire la miglior alimentazione per te
Ciò che ti si propone è un percorso piacevole e gratificante alla scoperta della tua salute, del gusto e del buon umore. Hai la possibilità di utilizzare uno strumento potentissimo, del tutto naturale come il cibo ed il movimento per migliorare la tua qualità di vita, perché rinunciarci?
Per ogni paziente si studia un percorso personale che esalti il buon umore, anche grazie al calcolo dei nutrienti che gli sono utili. Si favorisce, attraversano varie tappe, un processo naturale che riporta la persona in equilibrio nella sua globalità. Se sei triste o irritabile tenderai a mangiare sempre di più e abbandonare la dieta rapidamente. Se invece il percorso che segui ti fa sentire bene, in forma, pieno di energie, ti rende allegro, lo seguirai volentieri e otterrai dei risultati duraturi nel tempo.
Il ruolo del Biologo Nutrizionista
Il ruolo di Nutrizionista Cosenza Dott.ssa Chiara Palermo è principalmente quello di fare educazione alimentare, in modo che il paziente acquisisca una consapevolezza alimentare che gli permetta di mantenere “nel lungo periodo” gli obbiettivi prefissati e raggiunti.
Questa coscienza alimentare non significa soltanto dimagrire, ma raggiungere un’armonia con il cibo, che permetterà di gestire al meglio le proprie attività giornaliere.
“La salute non è semplicemente l’assenza di malattia, ma lo stato di completo benessere fisico, mentale e sociale”.
Educarsi ad una corretta alimentazione e migliorare il proprio stile di vita contribuisce, in maniera determinante, a migliorare la qualità di vita e la sua durata.
L’alimentazione svolge un ruolo fondamentale nello sviluppo del bambino e dell’adolescente, nell’equilibrio dell’adulto e nella longevità dell’anziano. Il nostro organismo necessita ogni giorno di tutti i nutrienti in quantità e rapporti ben definiti.
Per definire meglio queste quantità e questi rapporti è fondamentale conoscere il fabbisogno energetico del singolo individuo.
L’intervento di Nutrizionista Cosenza Dott.ssa Chiara Palermo
Il nutrizionista, attraverso la valutazione dei parametri antropometrici e del dispendio energetico (valutazione dello stato nutrizionale), interviene adottando:
- piani alimentari personalizzati equilibrate per raggiungere il peso ideale;
- piani alimentari personalizzati equilibrati e ipocalorici per perdere peso corporeo e raggiungere il peso ideale;
- regimi alimentari adeguati per pazienti con allergie ed intolleranze alimentari;
- regimi alimentari adeguati per pazienti con patologie dell’apparato digerente, respiratorio, osteoarticolare, patologie endocrine e metaboliche, del rene e delle vie urinarie, dermatologiche ed estetiche, ecc.
Inoltre il Nutrizionista interviene migliorando le abitudini alimentari con strategie di sorveglianza nutrizionale su pazienti in particolari condizioni fisiologiche quali:
- gravidanza
- allattamento
- crescita
- senescenza
- menopausa
- stati concomitanti come: osteoporosi, asma, perdita di capelli, tiroidite, nervosismo ecc.
Quali sono i requisiti fondamentali di un bravo nutrizionista?
- Il nutrizionista deve possedere una solida conoscenza delle proprietà dei nutrienti e dei non nutrienti contenuti negli alimenti e le eventuali modificazioni durante i processi tecnologici.
- Deve conoscere specificatamente i meccanismi biochimici e fisiologici della digestione e dell’assorbimento, i processi metabolici a carico dei nutrienti e riconoscere gli effetti dovuti alla malnutrizione per eccesso e per difetto.
- Deve essere capace di valutare la composizione corporea nei suoi sottoinsiemi fondamentali (molecole, cellule, tessuti) e le tecniche di valutazione dei singoli distretti; il metabolismo corporeo, il dispendio energetico, le tecniche ed i metodi di misura.
- Il nutrizionista deve essere in grado di conoscere e applicare le principali tecniche di laboratorio per valutare lo stato di nutrizione relativo ai macronutrienti e micronutrienti e saperne interpretare i risultati.
- È necessario che conosca la legislazione alimentare e sanitaria nazionale e comunitaria per quanto riguarda la commercializzazione e il controllo degli alimenti, degli ingredienti, degli additivi e degli integratori alimentari.
- Deve essere a conoscenza delle principali tecnologie industriali applicate nella preparazione di integratori alimentari e di alimenti destinati ad alimentazioni particolari.
- È indispensabile che sia in grado di definire gli apporti energetici e le qualità nutrizionali dei singoli alimenti e che conosca la composizione di base ed i fattori che regolano la biodisponibilità dei macro e micronutrienti.
- Inoltre è essenziale che conosca l’influenza degli alimenti sul benessere e sulla prevenzione delle malattie ed i livelli di sicurezza degli stessi sottoposti a trasformazioni tecnologiche e/o biotecnologiche, nonché i livelli tossicologici, le dosi giornaliere accettabili ed il rischio valutabile nell’assunzione di sostanze contenute o veicolate dalla dieta.
- E ancora il nutrizionista deve conoscere le tecniche di rilevamento dei consumi alimentari e le strategie di sorveglianza nutrizionale su popolazioni in particolari condizioni fisiologiche, quali gravidanza, allattamento, crescita, senescenza ed attività sportive.
- Poi è opportuno che conosca le problematiche relative alle politiche alimentari nazionali ed internazionali; deve saper valutare la qualità nutrizionale, la sicurezza, l’idoneità degli alimenti per il consumo umano, la malnutrizione in eccesso o in difetto nell’individuo e nelle popolazioni.
- Il nutrizionista deve essere in grado di analizzare la biodisponibilità dei nutrienti negli alimenti e negli integratori alimentari e i loro effetti; applicare metodiche atte a valutare la sicurezza degli alimenti e la loro idoneità per il consumo umano.
- Inoltre è fondamentale che sappia verificare la corretta assunzione di alimenti per raggiungere i livelli raccomandati di nutrienti per il mantenimento dello stato di salute.
- Deve saper valutare lo stato di nutrizione più consono alle caratteristiche fisiche e psichiche dell’individuo sottoposto a stress, con particolare riguardo all’attività fisica ed agonistica.
- Infine deve saper informare ed educare gli operatori istituzionali e la popolazione generale sui principi di sicurezza alimentare.
Rivolgendoti a Nutrizionista Cosenza Dott.ssa Chiara Palermo potrai essere centro di ritrovare tutte queste peculiarità per essere assolutamente certo di affidarti ad un professionista altamente qualificato e responsabile.
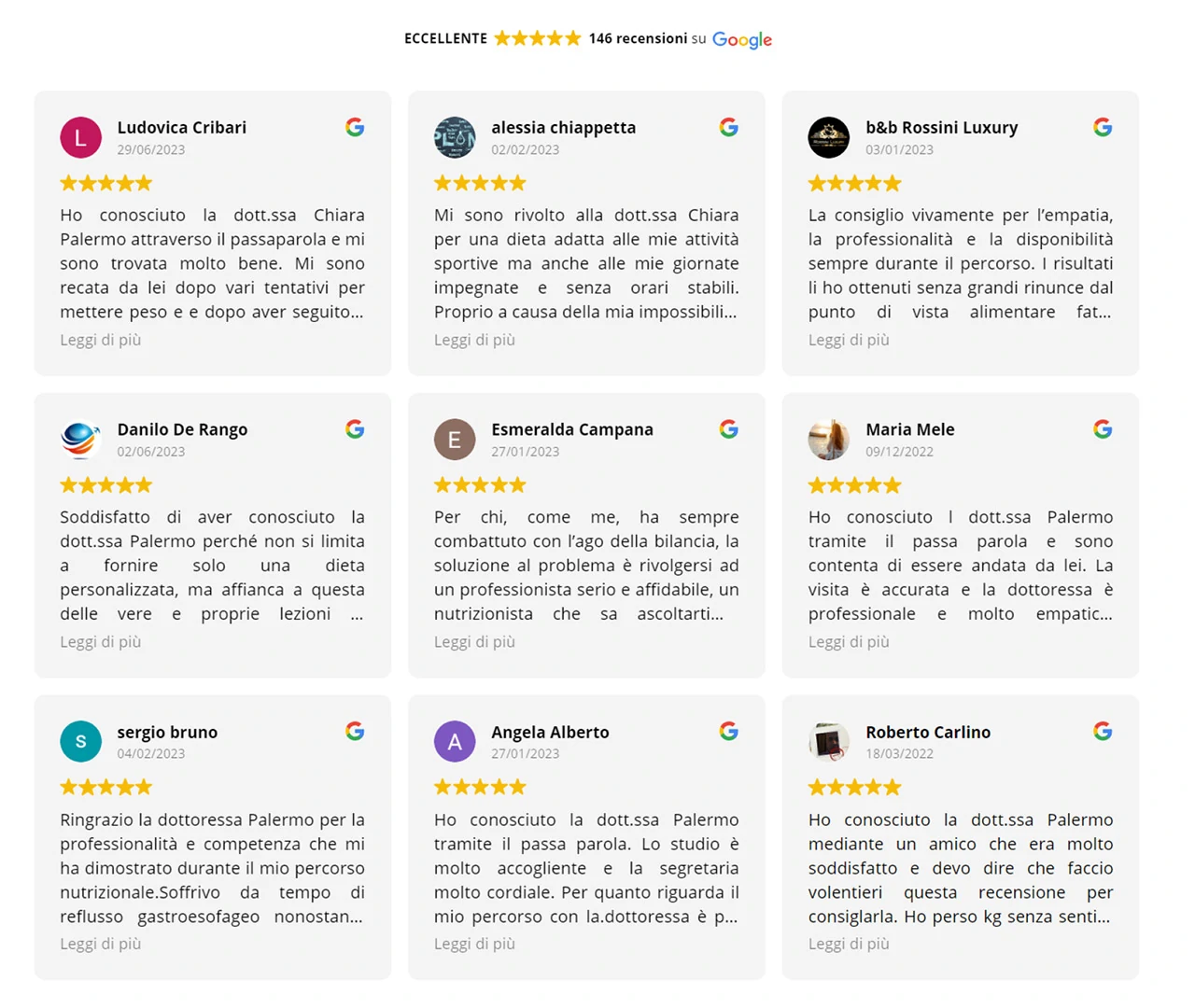
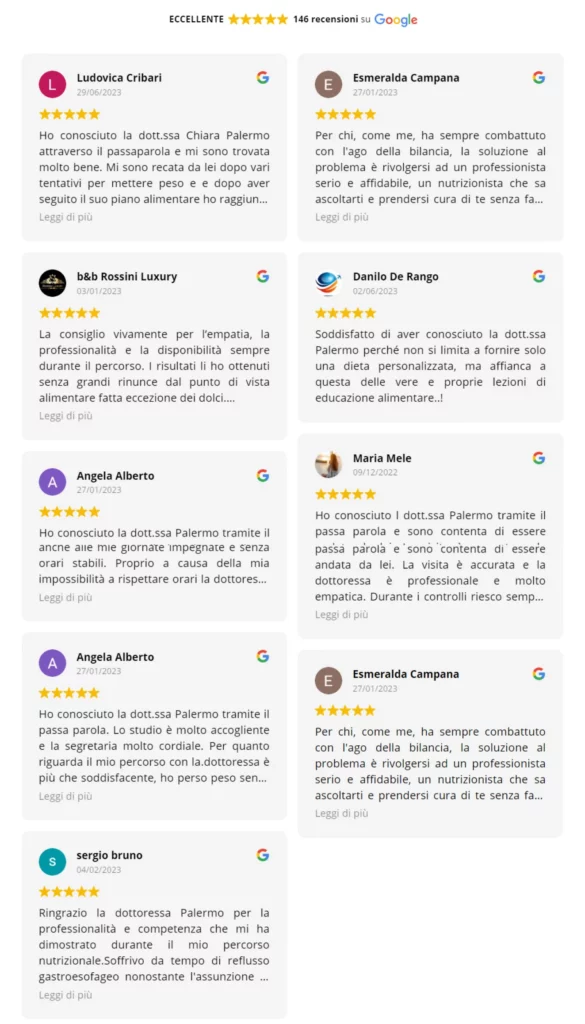
Dicono di me su Google
Contattami con fiducia
SCRIVI UN MESSAGGIO EMAIL ALLA DOTT.SSA CHIARA PALERMO
Ti risponderò al più presto possibile